|
Research Interests (host responses):
|
Research Interests (Cancer):
|

Chauhan NR, Kundu S, Chattopadhyay D, Bal R… Chauhan S*. Transgenic Mouse Models Support a Protective Role of Type I IFN Response in SARS-CoV-2 infection-related Lung Immunopathology and Neuroinvasion. Cell Reports, Cell Rep. 2023 Nov 28;42(11):113275.
Type I interferon (IFN-I) response is the first line of host defense against invading viruses. In the absence of definite mouse models, the role of IFN-I in SARS-CoV-2 infection remains perplexing. Here, we develop two mouse models, one with constitutively high IFN-I response (hACE2; Irgm1-/-) and the other with dampened IFN-I response (hACE2; Ifnar1-/-), to comprehend the role of IFN-I response. We report that hACE2; Irgm1-/- mice are resistant to lethal SARS-CoV-2 infection. In contrast, a severe SARS-CoV-2 infection along with immune cell infiltration, cytokine storm, and enhanced pathology is observed in the lungs and brain of hACE2; Ifnar1-/- mice. The hACE2; Irgm1-/-Ifnar1-/- double-knockout mice display loss of the protective phenotype observed in hACE2; Irgm1-/- mice, suggesting that heightened IFN-I response accounts for the observed immunity. Taking the results together, we demonstrate that IFN-I protects from lethal SARS-CoV-2 infection, and Irgm1 (IRGM) could be an excellent therapeutic target against SARS-CoV-2.
Type I interferon (IFN-I) response is the first line of host defense against invading viruses. In the absence of definite mouse models, the role of IFN-I in SARS-CoV-2 infection remains perplexing. Here, we develop two mouse models, one with constitutively high IFN-I response (hACE2; Irgm1-/-) and the other with dampened IFN-I response (hACE2; Ifnar1-/-), to comprehend the role of IFN-I response. We report that hACE2; Irgm1-/- mice are resistant to lethal SARS-CoV-2 infection. In contrast, a severe SARS-CoV-2 infection along with immune cell infiltration, cytokine storm, and enhanced pathology is observed in the lungs and brain of hACE2; Ifnar1-/- mice. The hACE2; Irgm1-/-Ifnar1-/- double-knockout mice display loss of the protective phenotype observed in hACE2; Irgm1-/- mice, suggesting that heightened IFN-I response accounts for the observed immunity. Taking the results together, we demonstrate that IFN-I protects from lethal SARS-CoV-2 infection, and Irgm1 (IRGM) could be an excellent therapeutic target against SARS-CoV-2.

Mehto S, Jena KK , Yadav R, Priyadarsini S, Samal P, Krishna S, Dhar K, Jain A, Chauhan NR, Murmu KC, Bal R, Sahu R, Jaiswal P, Sahoo BS, Patnaik S, Kufer TS, Rusten TE, Chauhan S, Prasad P, Chauhan S. Selective Autophagy of RIPosomes Maintains Innate Immune Homeostasis during Bacterial Infection. EMBO J. Dec 1;41(23):e111289. Cover Page Article
The NOD1/2-RIPK2 is a key cytosolic signaling complex that activates NF-κB pro-inflammatory response against invading pathogens. However, uncontrolled NF-κB signaling can cause tissue damage leading to chronic diseases. The mechanisms by which the NODs-RIPK2-NF-κB innate immune axis is activated and resolved remain poorly understood. Here, we demonstrate that bacterial infection induces the formation of endogenous RIPK2 oligomers (RIPosomes) that are self-assembling entities that coat the bacteria to induce NF-κB response. Next, we show that autophagy proteins IRGM and p62/SQSTM1 physically interact with NOD1/2, RIPK2 and RIPosomes to promote their selective autophagy and limit NF-κB activation. IRGM suppresses RIPK2-dependent pro-inflammatory programs induced by Shigella and Salmonella. Consistently, the therapeutic inhibition of RIPK2 ameliorates Shigella infection- and DSS-induced gut inflammation in Irgm1 KO mice. This study identifies a unique mechanism where the innate immune proteins and autophagy machinery are recruited together to the bacteria for defense as well as for maintaining immune homeostasis.
Cover Page: Volume 41 Issue 23, 1 December 2022
The cover art in Mandala style depicts that bacterial infection induces the formation of RIPosomes (RIPK2 oligomers) that coat the bacteria to induce NF-κB response. The autophagy proteins IRGM and p62 coordinate selective autophagy of RIPosomes to limit the NF-κB activation and hence maintain innate immune homeostasis. (Scientific illustration by Swati Chauhan and Suhani Chauhan, Institute of Life Sciences, Bhubaneswar)
The NOD1/2-RIPK2 is a key cytosolic signaling complex that activates NF-κB pro-inflammatory response against invading pathogens. However, uncontrolled NF-κB signaling can cause tissue damage leading to chronic diseases. The mechanisms by which the NODs-RIPK2-NF-κB innate immune axis is activated and resolved remain poorly understood. Here, we demonstrate that bacterial infection induces the formation of endogenous RIPK2 oligomers (RIPosomes) that are self-assembling entities that coat the bacteria to induce NF-κB response. Next, we show that autophagy proteins IRGM and p62/SQSTM1 physically interact with NOD1/2, RIPK2 and RIPosomes to promote their selective autophagy and limit NF-κB activation. IRGM suppresses RIPK2-dependent pro-inflammatory programs induced by Shigella and Salmonella. Consistently, the therapeutic inhibition of RIPK2 ameliorates Shigella infection- and DSS-induced gut inflammation in Irgm1 KO mice. This study identifies a unique mechanism where the innate immune proteins and autophagy machinery are recruited together to the bacteria for defense as well as for maintaining immune homeostasis.
Cover Page: Volume 41 Issue 23, 1 December 2022
The cover art in Mandala style depicts that bacterial infection induces the formation of RIPosomes (RIPK2 oligomers) that coat the bacteria to induce NF-κB response. The autophagy proteins IRGM and p62 coordinate selective autophagy of RIPosomes to limit the NF-κB activation and hence maintain innate immune homeostasis. (Scientific illustration by Swati Chauhan and Suhani Chauhan, Institute of Life Sciences, Bhubaneswar)

Nath P, Chauhan NR, Jena KK, Datey A, Kumar ND, Mehto S, De S, Nayak TK, Priyadarsini S, Rout K, Bal R, Murmu KC, Kalia M, Patnaik S, Prasad P, Reggiori F, Chattopadhyay S, Chauhan S. Inhibition of IRGM establishes a robust antiviral immune state to restrict pathogenic viruses. EMBO Rep. 2021 Sep 1:e52948. doi: 10.15252/embr.202152948. Cover Page Article
The type I interferon (IFN) response is the major host arsenal against invading viruses. IRGM is a negative regulator of IFN responses under basal conditions. However, the role of human IRGM during viral infection has remained unclear. In this study, we show that IRGM expression is increased upon viral infection. IFN responses induced by viral PAMPs are negatively regulated by IRGM. Conversely, IRGM depletion results in a robust induction of key viral restriction factors including IFITMs, APOBECs, SAMHD1, tetherin, viperin, and HERC5/6. Additionally, antiviral processes such as MHC-I antigen presentation and stress granule signaling are enhanced in IRGM-deficient cells, indicating a robust cell-intrinsic antiviral immune state. Consistently, IRGM-depleted cells are resistant to the infection with seven viruses from five different families, including Togaviridae, Herpesviridae, Flaviviverdae, Rhabdoviridae, and Coronaviridae. Moreover, we show that Irgm1 knockout mice are highly resistant to chikungunya virus (CHIKV) infection. Altogether, our work highlights IRGM as a broad therapeutic target to promote defense against a large number of human viruses, including SARS-CoV-2, CHIKV, and Zika virus.
Cover Page: Volume 22, Issue 11, 04 November 2021
The cover art in Mandala style depicts that the depletion of IRGM in a human cell (central mandala) establishes a strong innate immune state that can restrict (red stoppers) a large number of dreaded pathogenic viruses (smaller mandalas) including SARS‐CoV2, ZIKA, West Nile, and Chikungunya viruses. (Cover concept by Santosh Chauhan, Institute of Life Sciences; cover illustration by Swati Chauhan and Suhani Chauhan)
The type I interferon (IFN) response is the major host arsenal against invading viruses. IRGM is a negative regulator of IFN responses under basal conditions. However, the role of human IRGM during viral infection has remained unclear. In this study, we show that IRGM expression is increased upon viral infection. IFN responses induced by viral PAMPs are negatively regulated by IRGM. Conversely, IRGM depletion results in a robust induction of key viral restriction factors including IFITMs, APOBECs, SAMHD1, tetherin, viperin, and HERC5/6. Additionally, antiviral processes such as MHC-I antigen presentation and stress granule signaling are enhanced in IRGM-deficient cells, indicating a robust cell-intrinsic antiviral immune state. Consistently, IRGM-depleted cells are resistant to the infection with seven viruses from five different families, including Togaviridae, Herpesviridae, Flaviviverdae, Rhabdoviridae, and Coronaviridae. Moreover, we show that Irgm1 knockout mice are highly resistant to chikungunya virus (CHIKV) infection. Altogether, our work highlights IRGM as a broad therapeutic target to promote defense against a large number of human viruses, including SARS-CoV-2, CHIKV, and Zika virus.
Cover Page: Volume 22, Issue 11, 04 November 2021
The cover art in Mandala style depicts that the depletion of IRGM in a human cell (central mandala) establishes a strong innate immune state that can restrict (red stoppers) a large number of dreaded pathogenic viruses (smaller mandalas) including SARS‐CoV2, ZIKA, West Nile, and Chikungunya viruses. (Cover concept by Santosh Chauhan, Institute of Life Sciences; cover illustration by Swati Chauhan and Suhani Chauhan)

Kolapalli SP, Sahu R, Chauhan NR, Jena KK, Mehto S, Das SK, Jain A, Rout M, Dash R, Swain RK, Lee DY, Rusten TE, Chauhan S*, Chauhan S*. RNA binding RING E3-ligase DZIP3/hRUL138 is a novel driver of cell cycle and cancer progression by employing a unique mechanism to stabilize Cyclin D1.
Cancer Research, 2021 Jan 15;81(2):315-331. doi: 10.1158/0008-5472.CAN-20-1871 PMID: 33067265. * Corresponding author
DZIP3/hRUL138 is a poorly characterized RNA binding RING E3-ubiquitin ligase with functions in embryonic development. In this study, we demonstrated that DZIP3 is a crucial driver of cancer cell growth, migration, and invasion. In mice and zebrafish cancer models, DZIP3 promoted tumor growth and metastasis. In line with these results, DZIP3 was frequently overexpressed in several cancer types. Depletion of DZIP3 from cells resulted in reduced expression of Cyclin D1 and a subsequent G1 arrest and defect in cell growth. Mechanistically, DZIP3 utilized its two different domains to interact and stabilize Cyclin D1 both at mRNA and protein levels. Using an RNA-binding lysine-rich region, DZIP3 interacted with the AU-rich region in 3'UTR of Cyclin D1 mRNA and stabilized it. Using a RING E3-ligase domain, DZIP3 interacted and increased K63-linked ubiquitination of Cyclin D1 protein to stabilize it. Remarkably, DZIP3 interacted with, ubiquitinated, and stabilized Cyclin D1 predominantly in the G1 phase of the cell cycle where it is needed for cell cycle progression. In agreement with this, a strong positive correlation of mRNA expression between DZIP3 and Cyclin D1 in different cancer types was observed. Additionally, DZIP3 regulated several cell cycle proteins by modulating the Cyclin D1-E2F axes. Taken together, this study demonstrated for the first time that DZIP3 employs a unique two-pronged mechanism in its stabilization of Cyclin D1 to drive cell cycle and cancer progression.
Cover Page: During mitosis, Ki-67 (red) is necessary for the formation of the perichromosomal layer and prevents chromosomes from collapsing into a single chromatin mass. Thus, the cells undergoing mitosis can be easily recognized using Ki-67 immunostaining, as the nucleus (blue) gives a floral appearance. A significantly higher number of mitotic floral cells were observed in control tumors as compared with the DZIP3 knockdown tumors.
Cancer Research, 2021 Jan 15;81(2):315-331. doi: 10.1158/0008-5472.CAN-20-1871 PMID: 33067265. * Corresponding author
DZIP3/hRUL138 is a poorly characterized RNA binding RING E3-ubiquitin ligase with functions in embryonic development. In this study, we demonstrated that DZIP3 is a crucial driver of cancer cell growth, migration, and invasion. In mice and zebrafish cancer models, DZIP3 promoted tumor growth and metastasis. In line with these results, DZIP3 was frequently overexpressed in several cancer types. Depletion of DZIP3 from cells resulted in reduced expression of Cyclin D1 and a subsequent G1 arrest and defect in cell growth. Mechanistically, DZIP3 utilized its two different domains to interact and stabilize Cyclin D1 both at mRNA and protein levels. Using an RNA-binding lysine-rich region, DZIP3 interacted with the AU-rich region in 3'UTR of Cyclin D1 mRNA and stabilized it. Using a RING E3-ligase domain, DZIP3 interacted and increased K63-linked ubiquitination of Cyclin D1 protein to stabilize it. Remarkably, DZIP3 interacted with, ubiquitinated, and stabilized Cyclin D1 predominantly in the G1 phase of the cell cycle where it is needed for cell cycle progression. In agreement with this, a strong positive correlation of mRNA expression between DZIP3 and Cyclin D1 in different cancer types was observed. Additionally, DZIP3 regulated several cell cycle proteins by modulating the Cyclin D1-E2F axes. Taken together, this study demonstrated for the first time that DZIP3 employs a unique two-pronged mechanism in its stabilization of Cyclin D1 to drive cell cycle and cancer progression.
Cover Page: During mitosis, Ki-67 (red) is necessary for the formation of the perichromosomal layer and prevents chromosomes from collapsing into a single chromatin mass. Thus, the cells undergoing mitosis can be easily recognized using Ki-67 immunostaining, as the nucleus (blue) gives a floral appearance. A significantly higher number of mitotic floral cells were observed in control tumors as compared with the DZIP3 knockdown tumors.
|
|

Jena KK, Mehto S, Nath P, Chauhan NR, Sahu R, Dhar K, Das SK, Kolapalli SP, Murmu KC, Jain A, Krishna S, Sahoo BS, Chattopadhyay S, Rusten TE, Prasad P, Chauhan S, Chauhan S*. Autoimmunity gene IRGM suppresses cGAS-STING and RIG-I-MAVS signaling to control interferon response. EMBO Rep. 2020 Jul 27:e50051. doi: 10.15252/embr.202050051.
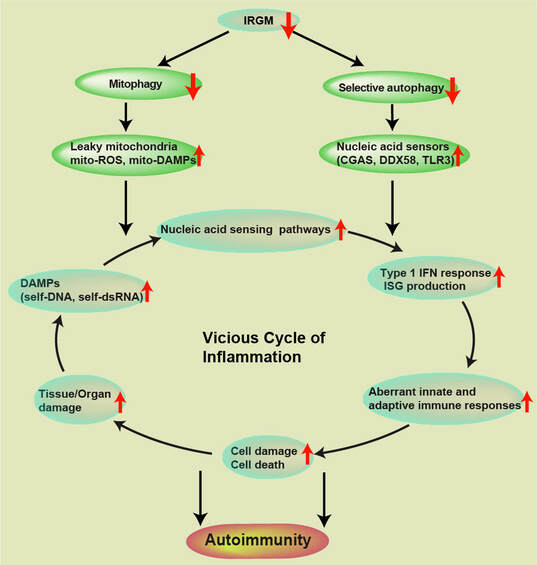
Our work published in EMBO Reports journal provides mechanistic insight of genesis and progression of autoimmune diseases. Interferon response over activation plays a significant role in the pathogenesis of several autoimmune diseases. We found that autophagy (and mitophagy) plays a significant role in the suppression of interferon response.
The type I interferon (IFN) response, where one side is first-line of defense against invading pathogens (esp. viruses including SARS-COV2), on the other side, its uncontrolled activation can lead to several autoimmune diseases. A fine homeostatic balance of type I IFN’s needs to be maintained to avoid autoimmune diseases. The knowledge of the master switches and the mechanisms that suppress the type I IFN response will be beneficial for generating therapeutics against autoimmune and viral diseases.
IRGM protein deficiency is genetically and functionally associated with several inflammatory and autoimmune diseases including ankylosing spondylitis, autoimmune thyroid diseases, Graves’ disease, Sjogren’s syndrome, Crohn’s disease, experimental autoimmune encephalomyelitis, hepatic steatosis, NAFLD and severe sepsis. The presence of IRGM in humans and mice is shown to be protective against autoimmune disorders. The connections between IRGM and several systemic autoimmune diseases argues a significant role of IRGM in innate immune homeostasis. The molecular mechanism by which human IRGM controls innate immune homeostasis in steady-state conditions and suppress autoimmunity remains completely undetermined.
This study uncovers that under homeostatic conditions, IRGM is a master suppressor of type I IFN response. The mRNA sequencing whole transcriptome analysis in human cells and mice shows that IRGM controls the expression of almost all major Interferon Stimulated Gene's. Mechanistically, we show that IRGM suppresses IFN signaling by mediating p62-dependent autophagic degradation of cGAS, RIG-I, and TLR3. Further, we found that IRGM is critical for the removal of damaged mitochondria by macroautophagy. Thus, IRGM deficiency results in defective mitophagy, accumulation of dysfunctional mitochondria, and enhanced mitochondrial DAMPs that stimulate cGAS-STING and RIG-I-MAVS axis to drive robust activation of type I IFN response leading to autoimmunity.
A prominent role of IRGM in suppressing inflammation presented in this study or previously by our lab (Mehto et al., 2019), justify why IRGM protein, which was dead for 20 million years of evolution, might have revived back in ancestors of humans. This study also defines IRGM as a strong potential target for new therapeutic interventions against autoimmune diseases and viral diseases.
The type I interferon (IFN) response, where one side is first-line of defense against invading pathogens (esp. viruses including SARS-COV2), on the other side, its uncontrolled activation can lead to several autoimmune diseases. A fine homeostatic balance of type I IFN’s needs to be maintained to avoid autoimmune diseases. The knowledge of the master switches and the mechanisms that suppress the type I IFN response will be beneficial for generating therapeutics against autoimmune and viral diseases.
IRGM protein deficiency is genetically and functionally associated with several inflammatory and autoimmune diseases including ankylosing spondylitis, autoimmune thyroid diseases, Graves’ disease, Sjogren’s syndrome, Crohn’s disease, experimental autoimmune encephalomyelitis, hepatic steatosis, NAFLD and severe sepsis. The presence of IRGM in humans and mice is shown to be protective against autoimmune disorders. The connections between IRGM and several systemic autoimmune diseases argues a significant role of IRGM in innate immune homeostasis. The molecular mechanism by which human IRGM controls innate immune homeostasis in steady-state conditions and suppress autoimmunity remains completely undetermined.
This study uncovers that under homeostatic conditions, IRGM is a master suppressor of type I IFN response. The mRNA sequencing whole transcriptome analysis in human cells and mice shows that IRGM controls the expression of almost all major Interferon Stimulated Gene's. Mechanistically, we show that IRGM suppresses IFN signaling by mediating p62-dependent autophagic degradation of cGAS, RIG-I, and TLR3. Further, we found that IRGM is critical for the removal of damaged mitochondria by macroautophagy. Thus, IRGM deficiency results in defective mitophagy, accumulation of dysfunctional mitochondria, and enhanced mitochondrial DAMPs that stimulate cGAS-STING and RIG-I-MAVS axis to drive robust activation of type I IFN response leading to autoimmunity.
A prominent role of IRGM in suppressing inflammation presented in this study or previously by our lab (Mehto et al., 2019), justify why IRGM protein, which was dead for 20 million years of evolution, might have revived back in ancestors of humans. This study also defines IRGM as a strong potential target for new therapeutic interventions against autoimmune diseases and viral diseases.

Mehto S, Jena KK, Nath P, Chauhan S, Kolapalli SP, Das SK, Sahoo PK, Jain A, Taylor GA, Chauhan S. The Crohn's disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Molecular Cell. 2019.Feb 7;73(3):429- 445.e7.
@Preview: Nabar NR, Kehrl JH. Inflammasome Inhibition Links IRGM to Innate Immunity. Mol Cell. 2019 Feb 7;73(3):391-392
@Preview: Nabar NR, Kehrl JH. Inflammasome Inhibition Links IRGM to Innate Immunity. Mol Cell. 2019 Feb 7;73(3):391-392
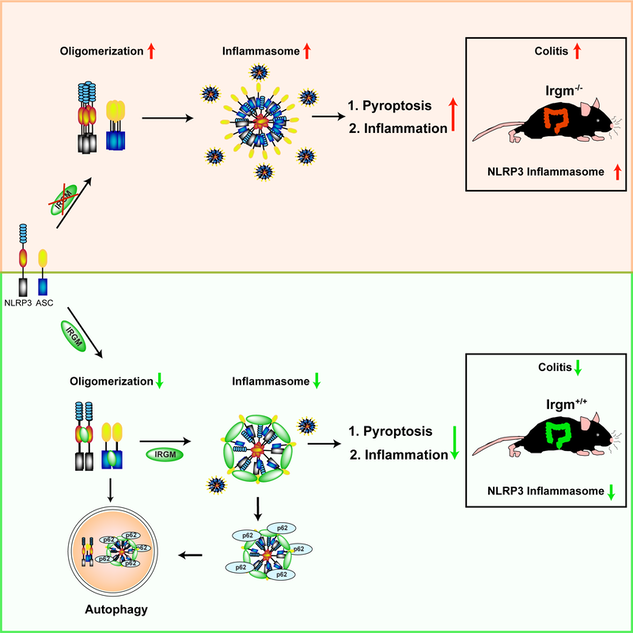
Title: Elucidation of a mechanism that combats autoimmune and autoinflammatory diseases in human
Aim: IRGM is genetically and functionally linked with protection against several chronic inflammatory diseases including inflammatory bowel disease (IBD), Crohn's disease (CD), leprosy, gastric cancer, chronic periodontitis, severe sepsis, and ankylosing spondylitis. The objective of this study is to define the mechanism by which IRGM is protective against these diseases.
Key finding(s): This study show that IRGM inhibits NLRP3 inflammasome activation to keep the inflammation under check. This study provides a basis for the protective function of IRGM in inflammatory and autoimmune diseases in human.
Strategy/Approach: The cell culture and animal models are used to discover the mechanism.
Results and Conclusion: This study defined a direct role of IRGM in suppressing NLRP3 inflammasome and inflammatory diseases related to NLRP3. We found that IRGM suppresses IL-1β production by limiting the activation of the NLRP3 inflammasome. IRGM limits inflammasome activation by two mechanisms, (1) limiting assembly of inflammasome components and (2) mediating the autophagic breakdown of inflammasome components. IRGM/Irgm1 can protect from inflammation-induced cell death and can suppress gut inflammation in Crohn’s disease experimental mice model.
Impact/Benefits: Mutations/dysregulation of NLRP3 have been linked with a range of auto-inflammatory and autoimmune diseases mostly characterized by increased production of IL-1β. So, for therapeutic purposes, it is of paramount importance to understand the mechanisms by which the cell restrains the activation of NLRP3 inflammasome and IL-1β production. This study define one of the important mechanisms by which IRGM suppress activation of NLRP3 inflammasome and IL-1β production. Currently, strategies (chemical and biological) are underway to increase IRGM activity so as to suppress inflammasomes for therapeutic purposes in inflammatory diseases. The strategies to target the IRGM-NLRP3 interaction as potential therapeutic interventions for IRGM- and NLRP3-associated diseases including Crohn’s is under consideration.
Aim: IRGM is genetically and functionally linked with protection against several chronic inflammatory diseases including inflammatory bowel disease (IBD), Crohn's disease (CD), leprosy, gastric cancer, chronic periodontitis, severe sepsis, and ankylosing spondylitis. The objective of this study is to define the mechanism by which IRGM is protective against these diseases.
Key finding(s): This study show that IRGM inhibits NLRP3 inflammasome activation to keep the inflammation under check. This study provides a basis for the protective function of IRGM in inflammatory and autoimmune diseases in human.
Strategy/Approach: The cell culture and animal models are used to discover the mechanism.
Results and Conclusion: This study defined a direct role of IRGM in suppressing NLRP3 inflammasome and inflammatory diseases related to NLRP3. We found that IRGM suppresses IL-1β production by limiting the activation of the NLRP3 inflammasome. IRGM limits inflammasome activation by two mechanisms, (1) limiting assembly of inflammasome components and (2) mediating the autophagic breakdown of inflammasome components. IRGM/Irgm1 can protect from inflammation-induced cell death and can suppress gut inflammation in Crohn’s disease experimental mice model.
Impact/Benefits: Mutations/dysregulation of NLRP3 have been linked with a range of auto-inflammatory and autoimmune diseases mostly characterized by increased production of IL-1β. So, for therapeutic purposes, it is of paramount importance to understand the mechanisms by which the cell restrains the activation of NLRP3 inflammasome and IL-1β production. This study define one of the important mechanisms by which IRGM suppress activation of NLRP3 inflammasome and IL-1β production. Currently, strategies (chemical and biological) are underway to increase IRGM activity so as to suppress inflammasomes for therapeutic purposes in inflammatory diseases. The strategies to target the IRGM-NLRP3 interaction as potential therapeutic interventions for IRGM- and NLRP3-associated diseases including Crohn’s is under consideration.

Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, Ahad A, Syed GH, Raghav SK, Senapati S, Chauhan S, Chauhan S. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018 Sep 14;37(18).
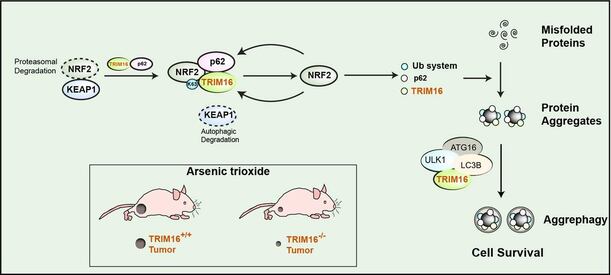
Laymen Title: Understanding the mechanism of safe disposal of stress-induced proteins aggregates for increased survival of body cell: implication in neurodegeneration and cancer.
Scientific Understanding
Misfolded proteins are a common outcome of protein biosynthesis in our body cell, and about 30% of newly synthesized proteins end up misfolded. Normally, these misfolded proteins are ubiquitinated and degraded by the proteasome system and/or autophagy process. Multiple factors ranging from genetic mutations (in neurodegeneration) to cellular (oxidative/proteotoxic stress) and environmental stresses can trigger protein misfolding overwhelming the cellular capacity to clear them leading to their aggregation and accumulation resulting in many pathological conditions collectively termed as proteinopathies (Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis, etc.).
On the flip side, a recent study showed that the augmented capacity to degrade the misfolded proteins alleviates oxidative stress associated with cancerous growth and is required for both the initiation and maintenance of malignant phenotypes (Chen et al. 2017, Cell reports). So the cancer cells hijack the machinery of protein quality control and utilizes them for their survival.
Given the paramount importance of process (in neurodegeneration and cancer) of formation of protein aggregates from misfolded proteins and their subsequent degradation, it is vital to understand the mechanism by which turnover of protein aggregates takes place in the cell. Till date, the mechanism by which protein aggregates are formed and degraded in cell remains elusive.
In this study, we discovered a novel mechanism by which cells during the oxidative or proteotoxic stress conditions can turn over the protein aggregates for their better survival. Two proteins namely, TRIM16 and NRF2 governs a protein machinery (p62 and ubiquitination) to regulate the process of protein aggregates formation and subsequently the TRIM16 engages autophagy machinery (a process of degradation in our cell) to degrade the protein aggregates. Thus, TRIM16 streamlines the process of stress-induced aggregate clearance and protects cells against oxidative/proteotoxic stress-induced toxicity. Thus, pharmacological targeting of TRIM16 for increasing its activity will be useful in neurodegeneration. On the flipside, we found that genetic knockout of this protein in cancer cells cripples their capability to handle oxidative stress or oncogenic stress leading to significant reduction in tumor size. Thus, pharmacological targeting of TRIM16 for decreasing its activity will be useful in killing cancer cells.
Laymen Understanding:
The formation of protein aggregates is a hallmark of neurodegeneration. The accumulation of protein aggregates due to incapability of neurons to clear them leads to cell death and is linked to pathological conditions in neurodegenerative diseases. On the flipside, cancer cells are smart enough to degrade stress-induce protein aggregates by hijacking the protein quality control machinery and survive in worse stress conditions (in which normal cell will die). Understanding the mechanism of protein aggregates turnover will pave path for therapeutic interventions of neurodegeneration and cancer. In this study, we defined a novel mechanism by which cells handle stress-induced protein aggregates turnover. TRIM16 uses two-pronged approach for complete turnover of protein aggregates under stress conditions. It governs the NRF2 mediated anti-oxidant system, ubiquitin mediated protein folding machinery and autophagy-mediated degradation machinery to accomplish this task. Hence therapeutic targeting of TRIM16 could be useful in neurodegeneration and cancer.
Scientific Understanding
Misfolded proteins are a common outcome of protein biosynthesis in our body cell, and about 30% of newly synthesized proteins end up misfolded. Normally, these misfolded proteins are ubiquitinated and degraded by the proteasome system and/or autophagy process. Multiple factors ranging from genetic mutations (in neurodegeneration) to cellular (oxidative/proteotoxic stress) and environmental stresses can trigger protein misfolding overwhelming the cellular capacity to clear them leading to their aggregation and accumulation resulting in many pathological conditions collectively termed as proteinopathies (Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis, etc.).
On the flip side, a recent study showed that the augmented capacity to degrade the misfolded proteins alleviates oxidative stress associated with cancerous growth and is required for both the initiation and maintenance of malignant phenotypes (Chen et al. 2017, Cell reports). So the cancer cells hijack the machinery of protein quality control and utilizes them for their survival.
Given the paramount importance of process (in neurodegeneration and cancer) of formation of protein aggregates from misfolded proteins and their subsequent degradation, it is vital to understand the mechanism by which turnover of protein aggregates takes place in the cell. Till date, the mechanism by which protein aggregates are formed and degraded in cell remains elusive.
In this study, we discovered a novel mechanism by which cells during the oxidative or proteotoxic stress conditions can turn over the protein aggregates for their better survival. Two proteins namely, TRIM16 and NRF2 governs a protein machinery (p62 and ubiquitination) to regulate the process of protein aggregates formation and subsequently the TRIM16 engages autophagy machinery (a process of degradation in our cell) to degrade the protein aggregates. Thus, TRIM16 streamlines the process of stress-induced aggregate clearance and protects cells against oxidative/proteotoxic stress-induced toxicity. Thus, pharmacological targeting of TRIM16 for increasing its activity will be useful in neurodegeneration. On the flipside, we found that genetic knockout of this protein in cancer cells cripples their capability to handle oxidative stress or oncogenic stress leading to significant reduction in tumor size. Thus, pharmacological targeting of TRIM16 for decreasing its activity will be useful in killing cancer cells.
Laymen Understanding:
The formation of protein aggregates is a hallmark of neurodegeneration. The accumulation of protein aggregates due to incapability of neurons to clear them leads to cell death and is linked to pathological conditions in neurodegenerative diseases. On the flipside, cancer cells are smart enough to degrade stress-induce protein aggregates by hijacking the protein quality control machinery and survive in worse stress conditions (in which normal cell will die). Understanding the mechanism of protein aggregates turnover will pave path for therapeutic interventions of neurodegeneration and cancer. In this study, we defined a novel mechanism by which cells handle stress-induced protein aggregates turnover. TRIM16 uses two-pronged approach for complete turnover of protein aggregates under stress conditions. It governs the NRF2 mediated anti-oxidant system, ubiquitin mediated protein folding machinery and autophagy-mediated degradation machinery to accomplish this task. Hence therapeutic targeting of TRIM16 could be useful in neurodegeneration and cancer.

Chauhan, S.*, Mandell, M. and Deretic, V.*. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Volume 58, Issue 3, p507–521, 7 May, 2015 Molecular Cell. * Corresponding author
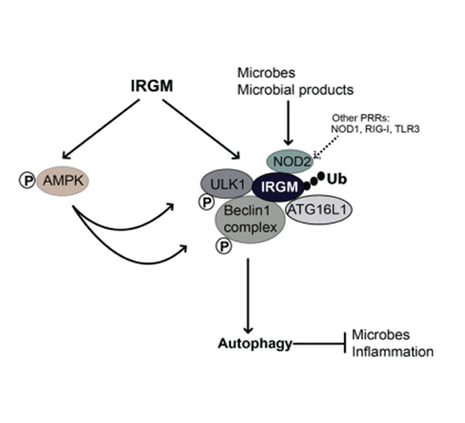
Polymorphisms in the IRGM gene, associated with Crohn disease (CD) and tuberculosis, are among the earliest identified examples documenting the role of autophagy in human disease. Functional studies have shown that IRGM protects against these diseases by modulating autophagy, yet the exact molecular mechanism of IRGM's activity has remained unknown. We have recently elucidated IRGM's mechanism of action. IRGM functions as a platform for assembling, stabilizing, and activating the core autophagic machinery, while at the same time physically coupling it to conventional innate immunity receptors. Exposure to microbial products or bacterial invasion increases IRGM expression, which leads to stabilization of AMPK. Specific protein-protein interactions and post-translational modifications such as ubiquitination of IRGM, lead to a co-assembly with IRGM of the key autophagy regulators ULK1 and BECN1 in their activated forms. IRGM physically interacts with two other CD risk factors, ATG16L1 and NOD2, placing these three principal players in CD within the same molecular complex. This explains how polymorphisms altering expression or function of any of the three factors individually can affect the same process - autophagy. Furthermore, IRGM's interaction with NOD2, and additional pattern recognition receptors such as NOD1, RIG-I and select TLRs, transduces microbial signals to the core autophagy apparatus. This work solves the long-standing enigma of how IRGM controls autophagy.

A punctum to this paper is published in Autophagy Journal.
DOI: 10.1080/15548627.2015.1084457
A punctum to this paper is published in Autophagy Journal.
DOI: 10.1080/15548627.2015.1084457

Chauhan, S., Goodwin, J.G., Chauhan, S., Manyam, G., Wang, J., Kamat, A.M., and Boyd, D.D. (2013). ZKSCAN3 Is a Master Transcriptional Repressor of Autophagy. Molecular Cell. Apr 11, 2013 ;50(1):16-2
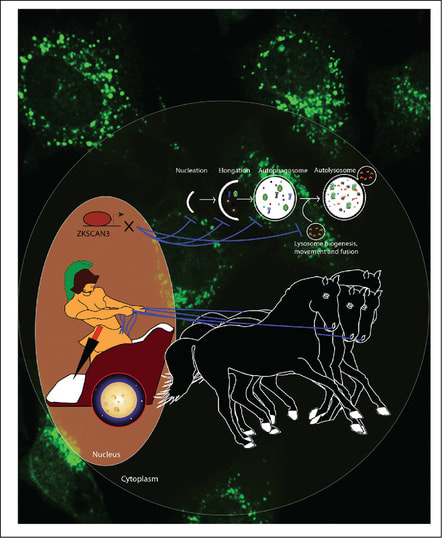
Autophagy constitutes a major cell-protective mechanism that eliminates damaged components and maintains energy homeostasis via recycling nutrients under normal/stressed conditions. Although the core components of autophagy have been well studied, regulation of autophagy at the transcriptional level is poorly understood. In this paper, we establish ZKSCAN3, a zinc finger family DNA-binding protein, as a transcriptional repressor of autophagy. Silencing of ZKSCAN3 induced autophagy and increased lysosome biogenesis. Importantly, we show that ZKSCAN3 represses transcription of a large gene set (>60) integral to, or regulatory for, autophagy and lysosome biogenesis/function and that a subset of these genes, including Map1lC3b and Wipi2, represent direct targets. Interestingly, ZKSCAN3 and TFEB are oppositely regulated by starvation and in turn oppositely regulate lysosomal biogenesis and autophagy, suggesting that they act in conjunction. Altogether, our study uncovers an autophagy master switch regulating the expression of a transcriptional network of genes integral to autophagy and lysosome biogenesis/function.

This paper was Highlighted in Cancer Discovery (AACR) journal, February 28, 2013; doi:10.1158/2159-8290.CD-RW2013-046. In this highlight the ZKSCAN3 is described as switch, controlling the autophagy and lysosome biogenesis.

This paper was also Recommended by Faculty of 1000 (http://f1000.com/prime/717980675)

Santosh Chauhan, Zahra Ahmed, Steven B. Bradfute,….Vincent Piguet, and Vojo Deretic. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nature communications, 2015 Oct 27;6:8620. doi: 10.1038/ncomms9620.
Autophagy is a conserved homeostatic process active in all human cells and affecting a spectrum of diseases. In this study, we use a pharmaceutical screen (>3000 FDA approved drugs) to discover new mechanisms for activation of autophagy. We identify a subset of pharmaceuticals inducing autophagic flux with effects in diverse cellular systems modelling specific stages of several human diseases such as HIV transmission and hyperphosphorylated tau accumulation in Alzheimer's disease. One drug, flubendazole, is a potent inducer of autophagy initiation and flux by affecting acetylated and dynamic microtubules in a reciprocal way. Disruption of dynamic microtubules by flubendazole results in mTOR deactivation and dissociation from lysosomes leading to Transcription Factor EB nuclear translocation and activation of autophagy. By inducing microtubule acetylation, flubendazole activates JNK1 leading to Bcl-2 phosphorylation, causing release of Beclin-1 from Bcl-2-Beclin-1 complexes for autophagy induction, thus uncovering a new approach to inducing autophagic flux that may be applicable in disease treatment.
Autophagy is a conserved homeostatic process active in all human cells and affecting a spectrum of diseases. In this study, we use a pharmaceutical screen (>3000 FDA approved drugs) to discover new mechanisms for activation of autophagy. We identify a subset of pharmaceuticals inducing autophagic flux with effects in diverse cellular systems modelling specific stages of several human diseases such as HIV transmission and hyperphosphorylated tau accumulation in Alzheimer's disease. One drug, flubendazole, is a potent inducer of autophagy initiation and flux by affecting acetylated and dynamic microtubules in a reciprocal way. Disruption of dynamic microtubules by flubendazole results in mTOR deactivation and dissociation from lysosomes leading to Transcription Factor EB nuclear translocation and activation of autophagy. By inducing microtubule acetylation, flubendazole activates JNK1 leading to Bcl-2 phosphorylation, causing release of Beclin-1 from Bcl-2-Beclin-1 complexes for autophagy induction, thus uncovering a new approach to inducing autophagic flux that may be applicable in disease treatment.
